
搜索结果: 1-15 共查到“理学 理论计算”相关记录130条 . 查询时间(0.278 秒)
中国化学会2020年软物质理论计算与模拟学术会议
软物质理论计算 2020年 学术会议
2023/2/23
中国化学会2020年软物质理论计算与模拟学术会议计划邀请在国内外从事软物质理论与模拟研究的学者进行一次跨学科的学术交流,以增进国内年轻学者对这一领域发展方向的认识,同时也为从事理论计算与模拟的学者提供一个展示和交流的平台,以此促进国内软物质理论计算与模拟研究方向的发展。
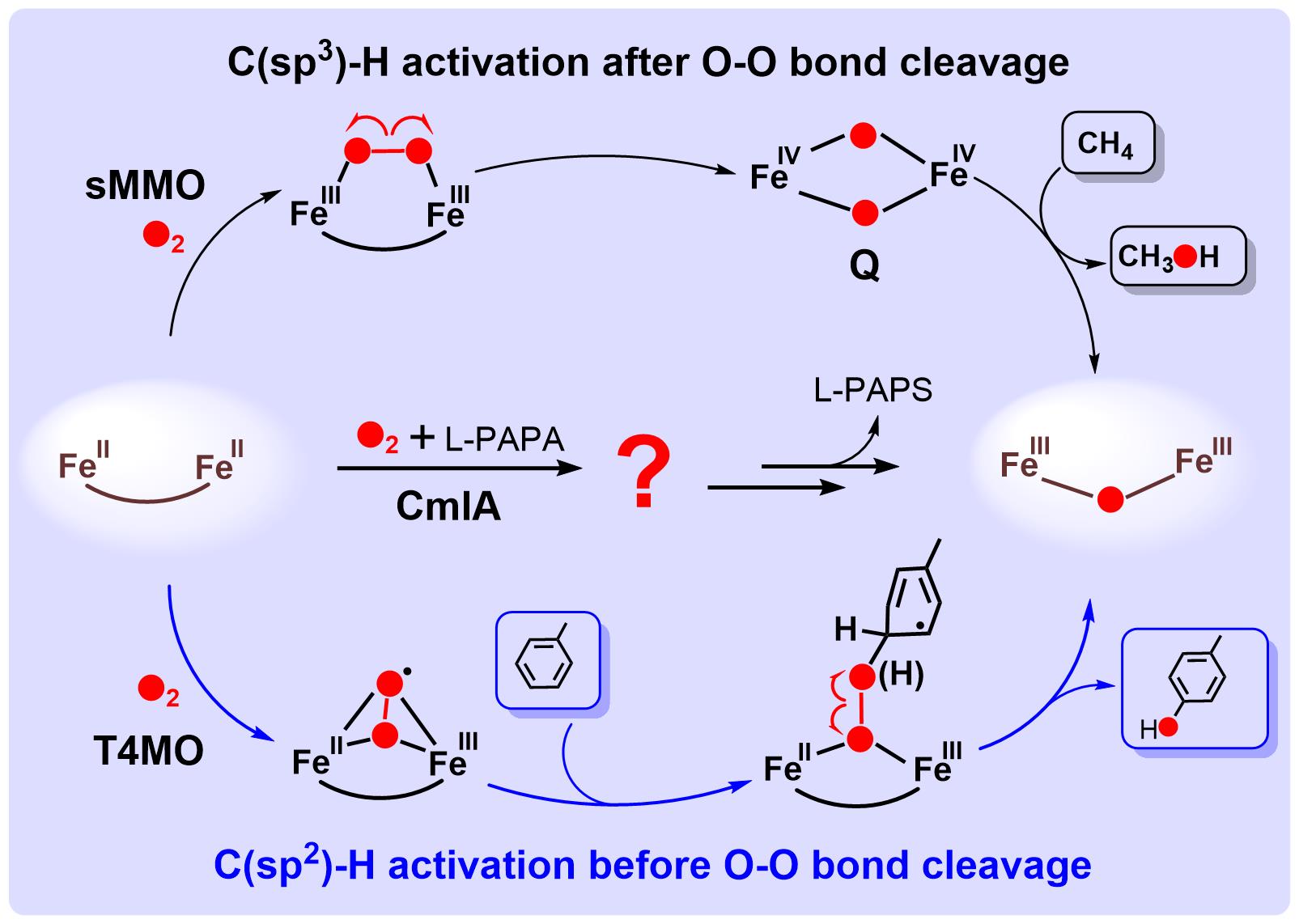
生物体内的C-H键羟化反应,普遍通过含铁加氧酶来实现。在羟化反应中,加氧酶将氧分子转化为具有底物羟化能力的含氧活性中间体实现O2的活化。对于双铁羟化酶,目前经典的O2活化机制源于可溶性甲烷单加氧酶(sMMO),其特点是O2活化后形成菱形双四价Fe2IVO2中间体(Q),接着通过Q抓氢过程,羟化甲烷中惰性C(sp3)-H键(图1),该O2活化机制常被用于推测其它双铁羟化酶的反应机制。然而,最近实验研...

陈辉团队通过理论计算研究提出过渡金属激发态反应性新概念(图)
陈辉 理论计算 过渡金属 激发态反应性
2023/1/6
不同于传统热化学反应,有机光催化可利用光能,在更温和的条件下驱动化学反应,该领域最近十余年取得了令人瞩目的进展。目前,光氧化还原(photo-redox)型有机光催化反应的发展,无论在实验还是机制方面,都已较为成熟,而过渡金属催化剂激发态直接诱导的光催化反应,其发展尚处于起步阶段。第一过渡系丰产金属,相比于第二、三过渡系的贵金属,具有较低的d轨道分裂能,其d-d型激发态能级较低,与可见光能量范围更...
以聚丙烯腈为原料, 利用静电纺丝技术和化学接枝制备得到硫脲基纳米螯合纤维, 并用于水溶液中 Cd(Ⅱ)的去除. 结合样品的表征和密度泛函(DFT)理论计算结果, 揭示了所制备纳米纤维材料对Cd(Ⅱ)的吸附机理. 借助静态吸附和动态吸附实验, 考察了硫脲基纳米螯合纤维对Cd(Ⅱ)的吸附性能. 结果表明, 纳米纤维吸附材料对Cd(Ⅱ)的最大吸附容量可达349.46 mg/g, 吸附过程在90 min以...
2020年11月14-15日,由中国人民大学数学学院和数学科学研究院主办的“蛋白质复合物和定量蛋白质组学:理论、计算与实验”研讨会在中国人民大学举办,会议得到中国计算机学会生物信息学专业委员会、中国运筹学会计算系统生物学分会的支持。为适应疫情防控常态化要求,研讨会报告采取线下线上相结合的方式,并在腾讯直播平台以及蔻享学术会议上直播。腾讯会议参与人数累计300余人,线上直播累计播放3000余次。

中国科学院金属研究所烷烃催化脱氢反应过程理论计算模拟研究获进展(图)
烷烃 催化脱氢 反应过程
2020/11/24
低碳烯烃是化工产业的支柱,是合成塑料、橡胶和纤维的基本原材料。烯烃产量是衡量一个国家化工产业能力重要指标之一,随着经济发展烯烃需求在持续增加,2020年我国烯烃消耗量占全球15%以上。由此可见,进一步提高烯烃生产效率有着重要经济价值和社会意义。另一方面,通过烷烃催化脱氢反应可以高效将低碳烷烃分子转化为同碳烯烃。目前,烷烃催化脱氢(直接和氧化)反应面临着选择性低、积碳以及高能耗等挑战,设计和开发新型...

近日,由国科大博士生导师、中科院精密测量科学与技术创新研究院研究员郑安民编著的专著《分子筛催化理论计算―从基础到应用》由科学出版社正式出版发行。分子筛催化剂在现代石油化工生产和环境治理领域有着广泛的应用,新型高效催化剂的设计依赖于对活性中心结构和反应中构型关系的揭示。理论计算化学作为实验科学的一种补充研究手段,能够在原子分子尺度确定分子筛催化剂的骨架结构、活性中心种类和反应机理,从而有助于更全面、...

近日,由精密测量院研究员郑安民编著的专著《分子筛催化理论计算―从基础到应用》由科学出版社正式出版发行。分子筛催化剂在现代石油化工生产和环境治理领域有着广泛的应用,新型高效催化剂的设计依赖于对活性中心结构和反应中构型关系的揭示。理论计算化学作为实验科学的一种补充研究手段,能够在原子分子尺度确定分子筛催化剂的骨架结构、活性中心种类和反应机理,从而有助于更全面、更深入地理解分子筛催化的本质。

下地幔及D”层矿物结构水的理论计算研究取得进展(图)
下地幔及D” 层矿物结构水
2019/9/3
中国科学院地球化学研究所矿床地球化学国家重点实验室博士研究生蒋佳俊与其导师、研究员张飞武,利用理论地球化学计算方法系统研究了水在布里奇曼石和后钙钛矿中的赋存机制,以及结构水对矿物相变和弹性性质的影响。研究发现:下地幔条件下,结构水对(Mg,Fe)SiO3矿物弹性波速和弹性模量(尤其是剪切波速和剪切模量)有着显著影响。矿物晶格中Si位取代机制中,当三价铁和0.55wt.%的氢共存时,模拟计算得到的布...

在国家自然科学基金委和中国科学院化学研究所理论计算化学平台项目的支持下,化学所光化学重点实验室研究员陈辉团队,在理论计算预测双核铁酶活性中间体结构及其反应机制研究方面取得系列进展。双核铁酶活化O2/NO的活性中间体结构,是揭示其自由基小分子活化机制的钥匙。然而,由于这些瞬态中间体的高活性和不稳定性,用传统的X-ray单晶衍射方法获取其结构是非常困难的。因此,如何通过其它方法预测双核铁酶含O2/NO...

近期,技术生物所黄青研究员课题组利用振动光谱和密度泛函理论计算解析灵芝酸,取得了新进展。

近日,由中国化学会主办,华东理工大学、中国化学会高分子学科委员会承办的2018年软物质理论计算与模拟学术会议在上海光大会展中心成功举办。来自全国66所高校和科研机构从事软物质理论研究的283位专家学者和研究生代表参加会议。中国科学院院士、我校副校长刘昌胜教授,中国科学院院士、复旦大学杨玉良教授,中国科学院院士、北京大学张平文教授,南京大学胡文兵教授,我校材料学院党委书记唐颂超教授等领导专家出席开幕...

Diels-Alder[4+2]环加成反应,是应用极其广泛的六元碳环合成方法。而对于更小的四元碳环的合成,根据Woodward-Hoffmann分子轨道对称守恒原理,协同的[2+2]环加成反应在基态时轨道对称性不匹配,将产生显著的动力学能垒,阻碍反应的发生。因此,相比于较成熟的[4+2]环加成反应,如何促使烯烃[2+2]环加成反应的进行,是有机合成方法学研究中的前沿领域之一。协同的烯烃[2+2]环...
为探索四聚吡咯配体和低价铀离子相互作用,以实验合成单层三明治结构配合物PcUⅥPc(Pc=酞菁)为基础,设计双层三明治型PzUmPzUmPz(m=Ⅲ,Ⅳ,Pz=氮杂卟啉),采用相对论密度泛函理论考察了其几何结构、异构体相对稳定性以及成键和轨道性质.得到se(staggered-eclipsed)和es(eclipsed-staggered)2种类型稳定空间异构体,
MnCl2/DMSO溶液的拉曼光谱和理论计算
氯化锰/二甲基亚砜溶液 拉曼光谱 溶剂化作用 溶剂化构型
2019/1/18
利用拉曼光谱研究了不同温度和浓度MnCl2/DMSO溶液体系离子的溶剂化作用,结果表明,在0~0.8 mol/L浓度范围内,随着浓度增加,Mn2+与DMSO的相互作用逐渐增强,S=O伸缩振动峰向低波数移动,S=O双键减弱;C-S伸缩振动峰向高波数移动,C-S键增强.温度升高,S=O双键和C-S键伸缩振动峰均向相反的方向移动,溶剂化作用减弱.56℃以上,单体DMSO迅速增加,与Mn2+溶剂化作用的D...