
搜索结果: 1-7 共查到“物理学 第一性原理”相关记录7条 . 查询时间(0.403 秒)
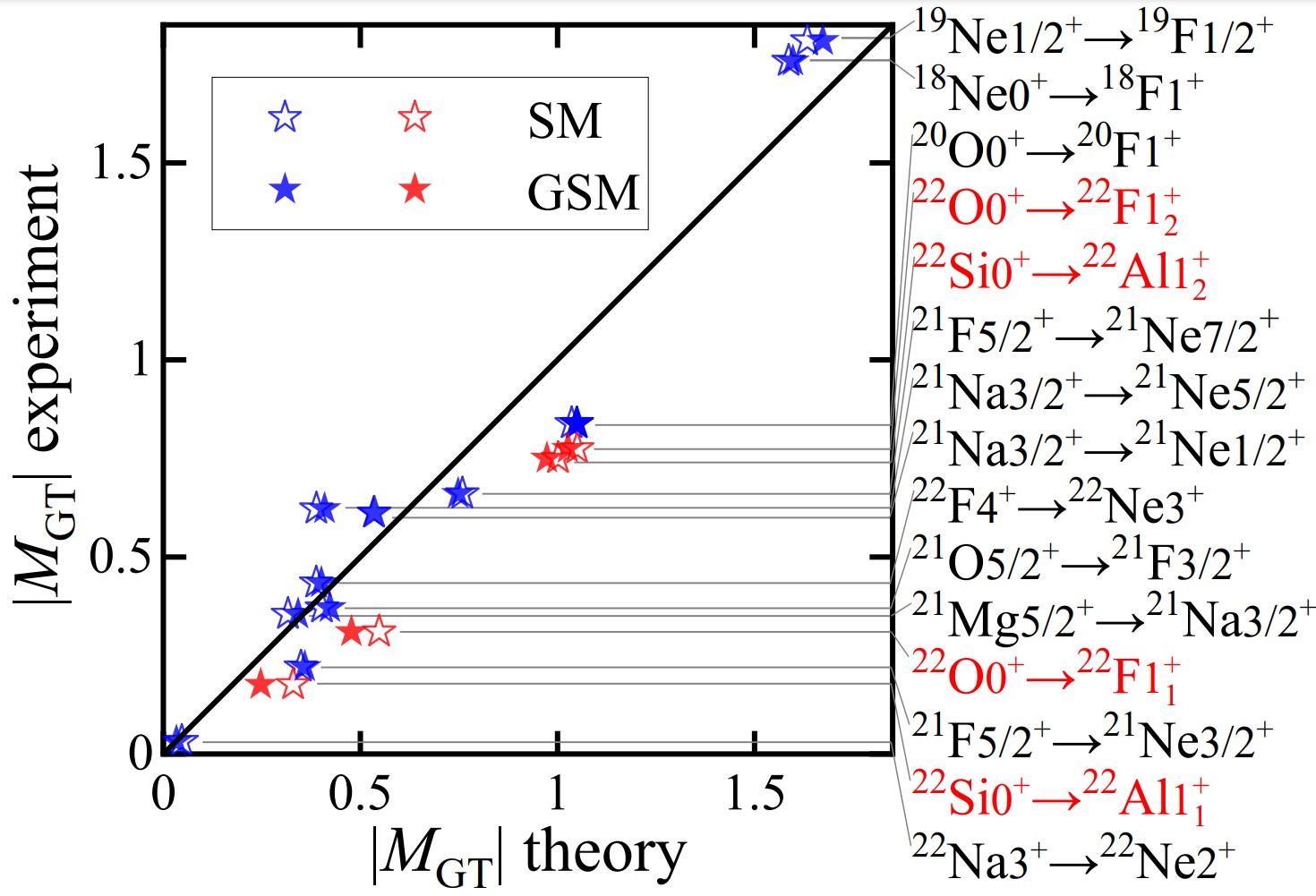
北京大学物理学院技术物理系、核物理与核技术国家重点实验室许甫荣教授团队发展了第一性原理的多体微扰理论框架下的复动量空间有效算符理论,首次将共振与连续态耦合包含在价空间有效算符的计算中,能自洽地描述滴线区不稳定原子核的能谱、β衰变及其它可观测量。

北京大学物理学院技术物理系、核物理与核技术国家重点实验室许甫荣教授团队发展了第一性原理的多体微扰理论框架下的复动量空间有效算符理论,首次将共振与连续态耦合包含在价空间有效算符的计算中,能自洽地描述滴线区不稳定原子核的能谱、β衰变及其它可观测量。

第一性原理计算已被广泛应用于物理、材料、化学、生物相关的科学研究。然而,受限于计算效率和精度,如何实现大尺度材料体系的第一性原理研究是该领域的一个重大挑战。基于人工神经网络的深度学习方法为解决该挑战问题带来了曙光。近期,深度学习已经成功应用于精确预测原子间相互作用,并加速分子动力学模拟。相比之下,理解、预测材料物性离不开电子结构计算,其深度学习的方法实现更具挑战性,研究进展有限。因此,发展深度学习...
石墨烯过渡层对金属/SiC接触肖特基势垒调控的第一性原理研究
SiC 石墨烯 肖特基势垒 第一性原理计算
2022/3/31
由于SiC禁带宽度大,在金属/SiC接触界面难以形成较低的势垒,制备良好的欧姆接触是目前SiC器件研制中的关键技术难题,因此,研究如何降低金属/SiC接触界面的肖特基势垒高度(SBH)非常重要。本文基于密度泛函理论的第一性原理赝势平面波方法,结合平均静电势和局域态密度计算方法,研究了石墨烯作为过渡层对不同金属(Ag,Ti,Cu,Pd,Ni,Pt)/SiC接触的SBH的影响。计算结果表明,单层石墨烯...
铁原子吸附联苯烯单层电子结构的第一性原理
联苯烯单层 吸附 电子结构 第一性原理计算
2022/3/17
联苯烯单层由碳原子的四元、六元和八元环组成,具有与石墨烯相似的单原子层结构.2021年5月,Science首次报道了该材料的实验合成,引起了科研工作者的极大关注.基于第一性原理的密度泛函方法,研究了铁原子在联苯烯单层的吸附构型并分析了其电子结构.结构优化、吸附能和分子动力学的计算表明,联苯烯单层的四元环空位是铁原子最稳定的吸附位点,吸附能可达1.56eV.电子态密度计算表明铁3d电子与碳的2p电子...
由于在磁性材料体系中缺失时间反演对称性,导致nodalchain被破坏,所以nodalchain通常存在于非磁材料中。但是,磁性材料EuAuBi是与常规磁性材料不同。本工作以第一性原理计算为研究方法,预言了在不考虑自旋轨道相互作用时,磁性材料EuAuBi体系为新型拓扑nodalchain半金属;当考虑自旋轨道耦合时,EuAuBi会退化为外尔半金属。对于非磁材料BaAuBi来说,在不考虑自旋轨道相互...